导师简介
如果你想申请香港浸会大学 计算机科学系博士,那今天这期文章解析可能对你有用!今天Mason学长为大详细解析香港浸会大学的Prof.Zhang的研究领域和代表文章,同时,我们也推出了新的内容“科研想法&开题立意”,为同学们的科研规划提供一些参考,并且会对如何申请该导师提出实用的建议!方便大家进行套磁!后续我们也将陆续解析其他大学和专业的导师,欢迎大家关注!
PhD导师简介 (480)")
教授现任香港浸会大学计算机科学系副教授。在加入香港浸会大学之前,导师曾在斯坦福大学计算机科学系和病理学系担任博士后研究员。导师于2012年获得香港大学李嘉诚医学院医学哲学硕士学位,并于2016年获得香港城市大学计算机科学博士学位。他于2008年获得天津大学软件工程学士学位。2015年,导师曾作为访问学者在加州大学伯克利分校数学系与Stephen Smale教授合作研究。导师拥有基因组学、统计学和计算机科学的跨学科背景。
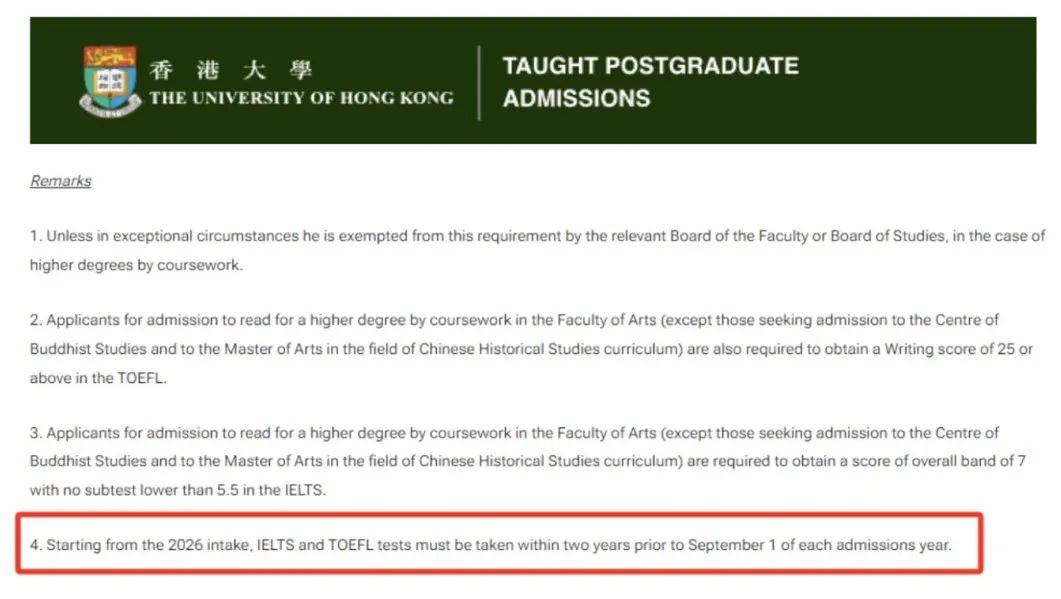
研究领域
导师的教学领域主要涵盖计算基因组学、生物信息学、人工智能在科学中的应用等方向。具体研究兴趣包括:
- 计算基因组学(Computational Genomics)
- 科学人工智能(AI for Science)
- 基因组学深度学习(Deep learning in Genomics)
- 单细胞多组学测序(Single-Cell Multiomics Sequencing)
- 基础模型(Foundation model)
- 大型语言模型(Large language model)
研究分析
1. stDyer enables spatial domain clustering with dynamic graph embedding
发表在《Genome Biology》,2025年
该研究提出了一种名为stDyer的创新计算方法,通过动态图嵌入技术实现空间域聚类。在空间转录组学快速发展的背景下,stDyer能够有效捕获空间域内基因表达的动态变化模式,为理解组织中复杂的细胞空间排布和功能提供了新工具。该方法整合了空间信息和基因表达数据,通过动态图嵌入技术实现对空间域的精确聚类,显著提高了识别功能上相似但空间上分散的细胞群的能力。
2. Deepurify: a multi-modal deep language model to remove contamination from metagenome-assembled genomes
发表在《Nature Machine Intelligence》,2024年
该研究提出了一种名为Deepurify的多模态深度语言模型,用于清除宏基因组组装基因组(MAGs)中的污染序列。在宏基因组分析中,污染是一个普遍存在的问题,会影响后续分析的准确性。Deepurify通过将基因组序列视为"语言",利用自然语言处理技术检测不同物种之间的序列差异,实现了对污染序列的高效识别和清除。
3. Exploring high-quality microbial genomes by assembling short-reads with long-range connectivity
发表在《Nature Communications》,2024年
该研究探索了通过组装具有远程连接性的短读序列来获取高质量微生物基因组的新方法。传统的短读测序虽然经济高效,但在复杂区域的组装方面存在挑战。该研究提出了一种创新的计算框架,通过整合短读序列和长距离连接信息,实现了对微生物基因组的高质量组装。研究团队应用该方法成功组装了多个高质量的微生物基因组,并发现了传统方法难以捕获的基因组结构变异。
4. dynDeepDRIM: a dynamic deep learning model to infer direct regulatory interactions using single cell time-course gene expression data
发表在《Briefings in Bioinformatics》,2022年
该研究提出了一种名为dynDeepDRIM的动态深度学习模型,用于推断单细胞时间序列基因表达数据中的直接调控相互作用。理解基因调控网络对揭示细胞功能和疾病机制至关重要,而传统方法难以从单细胞时间序列数据中准确推断动态调控关系。dynDeepDRIM通过深度学习架构捕获基因表达的时间动态特征,实现了对直接调控关系的高精度预测。
5. DeepDRIM: a deep neural network to reconstruct cell-type-specific gene regulatory network using single-cell RNA-Seq Data
这篇发表在《Briefings in Bioinformatics》,2021年
该研究提出了一种名为DeepDRIM的深度神经网络,用于使用单细胞RNA测序数据重建细胞类型特异性基因调控网络。单细胞RNA测序技术的发展使得研究者能够在单细胞水平研究基因表达,但如何从这些数据中构建准确的基因调控网络仍是一个挑战。DeepDRIM通过深度学习方法有效整合了基因表达模式和调控因子的信息,实现了对细胞类型特异性调控网络的重建。
6. Aquila enables reference-assisted diploid personal genome assembly and comprehensive variant detection based on linked reads
发表在《Nature Communications》
该研究开发了一种名为Aquila的工具,用于基于链接读序列进行参考辅助的二倍体个人基因组组装和全面变异检测。人类基因组的二倍体特性给基因组组装和变异检测带来了挑战,特别是对于复杂的结构变异。Aquila通过利用链接读序列技术(如10x Genomics)提供的长距离连接信息,实现了对人类二倍体基因组的高质量从头组装。该方法能够准确检测各种类型的变异,包括单核苷酸多态性、小型插入缺失和大型结构变异。
项目分析
1.人类肠道宏基因组高通量测序技术的计算工具开发
导师领导的这一研究项目旨在开发先进的计算工具,支持人类肠道宏基因组的高通量测序技术。在该项目中,导师团队开发了一系列创新的计算方法和工具,包括用于宏基因组组装的链接读序列技术应用和用于去除宏基因组组装基因 组中污染的深度学习模型,Deepurify这些工具极大地提高了肠道宏基因组分析的准确性和效率,为后续的微生物功能研究和与疾病关联的研究奠定了坚实基础。
2. 宏基因组学和单细胞多组学数据的基础模型开发
这一项目致力于为宏基因组学和单细胞多组学数据开发强大的基础模型。导师团队在该项目中借鉴自然语言处理领域的基础模型思想,将基因组序列视为特殊的"语言",开发了适用于基因组学数据的基础模型。这些基础模型为复杂生物学数据的分析提供了新范式,显著推动了多组学数据整合分析的能力,为精准医学研究提供了重要技术支持。
3.大型语言模型在现代医疗保健问题中的应用探索
该项目探索大型语言模型(LLM)在现代医疗保健问题中的创新应用。导师团队在该项目中探索如何将LLM技术应用于医疗数据分析、疾病诊断、治疗方案推荐等方面。团队还探索了LLM在医学文献挖掘、电子健康记录分析、医患互动等方面的应用,开发了多个基于LLM的医疗辅助系统原型。
研究想法
1. 多模态基础模型整合空间转录组学与单细胞多组学数据
基于导师在stDyer和DeepDRIM系列工作的基础上,可以探索开发一个多模态基础模型,同时整合空间转录组学和单细胞多组学数据。当前单细胞多组学研究与空间转录组学研究往往相对独立,但两种技术各有优势:单细胞多组学提供了细胞水平的高分辨率分子特征,而空间转录组学则保留了细胞的空间位置信息。通过构建一个统一的多模态基础模型,可以实现这两类数据的无缝整合,从而更全面地理解细胞状态、空间组织和功能关系。
2. 基于因果推理增强的基因调控网络重建框架
在导师DeepDRIM和dynDeepDRIM工作的基础上,可以引入因果推理机制,开发基于因果推理增强的基因调控网络重建框架。当前的基因调控网络重建主要基于相关性或机器学习方法,难以区分直接因果关系和间接相关关系。通过整合因果发现算法(如PC算法、FCI算法)与深度学习模型,可以更准确地推断基因之间的因果关系。
3. 自监督学习驱动的宏基因组长读序列分析框架
基于导师在宏基因组学研究的丰富经验,特别是Deepurify和链接读序列技术的应用,可以探索开发自监督学习驱动的宏基因组长读序列分析框架。随着三代测序技术(如PacBio和Oxford Nanopore)的快速发展,长读序列在宏基因组研究中的应用日益广泛,但长读序列的高错误率和宏基因组的复杂性给数据分析带来了挑战。通过自监督学习方法,可以有效利用未标记的大量宏基因组数据进行模型预训练,从而提高下游任务的性能。
4. 大型语言模型驱动的精准药物重定位系统
借鉴导师在将大型语言模型应用于医疗保健问题的研究方向,可以开发一个大型语言模型驱动的精准药物重定位系统。药物重定位(寻找已有药物的新适应症)是一种高效的药物开发策略,但传统方法往往依赖于有限的数据类型和模式。通过利用大型语言模型的强大语义理解和知识融合能力,可以整合多源异构数据(文献、电子健康记录、基因组数据、蛋白质结构、药物化学特性等),构建更全面的药物-疾病-靶点关系网络。
5. 时空多组学数据的动态网络建模与分析
在导师dynDeepDRIM对时间序列单细胞数据分析和stDyer对空间转录组学数据分析的基础上,可以探索时空多组学数据的动态网络建模与分析方法。生物系统本质上是动态的,细胞状态和相互作用随时间和空间不断变化,特别是在发育和疾病进程中。通过整合时间序列和空间信息的多组学数据,构建时空动态网络模型,可以更全面地捕获生物系统的动态变化特征。
申请建议
1. 学术背景准备
- 鉴于导师研究领域的特点,理想的学术背景应包括计算机科学(特别是机器学习、深度学习)和生物信息学/基因组学的复合背景。
- 如果你的本科或硕士阶段主修计算机科学,建议系统学习分子生物学、基因组学和统计学的基础知识;
- 如果主修生物学相关专业,则需要加强编程能力和算法设计思维,特别是Python、R等数据分析语言和TensorFlow、PyTorch等深度学习框架的应用能力。
2 研究经验与技能培养
理想的候选人应至少参与过一个与计算基因组学或生物信息学相关的研究项目,并有一定的成果(如会议论文、期刊论文或技术报告)。
特别重要的是,你应该能够展示以下几点能力:
- 独立数据分析能力:能够从原始测序数据出发,完成数据清洗、质量控制、组装/比对、注释和下游分析的完整流程
- 算法开发能力:能够根据生物学问题设计和实现适当的算法,尤其是机器学习模型的设计与训练
- 大数据处理能力:熟悉高性能计算平台(如集群、云计算)和常用的生物信息学工具
- 跨学科沟通能力:能够理解生物学问题并将其转化为可计算的模型
3 针对导师研究方向的个性化准备
- 深入研读导师论文:针对导师的核心研究方向(如宏基因组学、单细胞多组学分析、大型语言模型在基因组学中的应用),各选取2-3篇代表性论文进行深入分析。
- 复现关键算法:尝试复现导师论文中的部分算法或方法。例如,可以基于公开数据集实现DeepDRIM或Deepurify的简化版本,或者尝试使用导师开发的工具(如Aquila)分析公开的基因组数据。
- 思考创新点和改进空间:基于对导师研究的理解,提出有见地的问题和可能的改进方向。例如,如何将最新的大型语言模型技术应用于宏基因组分析?如何整合多模态组学数据提高基因调控网络重建的准确性?
- 关注领域前沿进展:定期关注计算基因组学和AI for Science领域的最新进展,包括顶级期刊(如Nature Methods、Genome Biology)的最新论文和重要会议(如RECOMB、ISMB)的报告。
博士背景
Aurelia ,美国TOP10院校计算机科学与认知科学双博士生,研究聚焦算法博弈论不确定性及其在人工智能中的应用。她的跨学科研究融合了计算机科学、语言学和心理学知识,在国际顶级期刊《Journal of Artificial Intelligence Research》和《Cognitive Science》上发表多篇论文。Aurelia 荣获ACM SIGAI博士论文奖,擅长相关方向的PhD申请指导。
【竞赛报名/项目咨询+微信:mollywei007】